It is a routine job to put 20 (or sometimes 30 or more) NMR structures calculated by Xplor-NIH or CYANA to a single ensemble file. The ensemble PDB separates the 20 coordinates by the MODEL/ENDMDL session (see my 2021 post for details). For example:
MODEL 1
ATOM ..
ATOM …
ENDMDL
MODEL 2
ATOM ..
ATOM ..
ENDMDL
We often selected the 20 lowest energy structures and combine them. However, simply putting all of them into one ensemble file with MODEL/ENMDL separation doesn’t present the structural convergence. Users have to manually do alignment, save as 20 new PDB files, and assemble the aligned PDB files. PDBstat can perform this job easily.
Here is a python script calling PyMOL to perform a similar task. The script was completely generated by chatGPT with my input of request functions.
This script utilizes PyMOL and it tests and runs in Ubuntu 24. it does the following jobs
- Load 20 PDB files listed in the “List_best20” text file.
- Generate an average PDB from the 20 coordinates, save it as “average.pdb”
- Run backbone alignment (CA, C, O, and N) one by one to the average structure, report the RMSD. The script interactively asks user to define the range of sequence to be aligned, for example 6-77.
- At the same time, the aligned coordinates will be saved together with MODEL/ENDMDL separations. The default saved name is “aligned_and_ensemble.pdb”.
- Then it does another two alignments to report the overall RMSD values for all residues (backbone), and overall RMSD for all atoms. The values are saved in a file called rmsd_results.txt.
import pymol
from pymol import cmd
import numpy as np
def generate_average_structure(list_file, output_file):
"""
Generate an average structure from a list of PDB files.
Parameters:
list_file (str): Path to the list file containing structure file paths.
output_file (str): Path to save the average structure as a PDB file.
"""
# Initialize PyMOL without GUI
pymol.finish_launching(['pymol', '-qc'])
# Read the list of PDB files
with open(list_file, 'r') as f:
structure_files = [line.strip() for line in f if line.strip()]
if not structure_files:
print("Error: No structures found in the list file.")
return
# Load structures and collect coordinates
coordinates = []
for i, structure_file in enumerate(structure_files):
structure_name = f'structure_{i+1}'
cmd.load(structure_file, structure_name)
coords = cmd.get_coords(f'{structure_name}')
if coords is not None:
coordinates.append(coords)
else:
print(f"Warning: No coordinates found in {structure_file}.")
if not coordinates:
print("Error: No valid coordinates collected.")
return
# Compute the average coordinates
avg_coords = np.mean(np.array(coordinates), axis=0)
# Create a new object for the average structure
cmd.load(structure_files[0], 'average_structure') # Use the first structure as a template
for i, coord in enumerate(avg_coords):
x, y, z = coord
cmd.alter_state(1, f'average_structure and index {i+1}', f"x={x:.6f}; y={y:.6f}; z={z:.6f}")
cmd.save(output_file, 'average_structure')
print(f"Average structure saved as {output_file}")
def align_to_average(list_file, average_file, output_ensemble):
"""
Align structures to an average structure and save as an ensemble.
Parameters:
list_file (str): Path to the list file containing structure file paths.
average_file (str): Path to the average structure PDB file.
output_ensemble (str): Path to save the aligned ensemble as a PDB file.
"""
# Initialize PyMOL without GUI
pymol.finish_launching(['pymol', '-qc'])
# Read the list of PDB files
with open(list_file, 'r') as f:
structure_files = [line.strip() for line in f if line.strip()]
if not structure_files:
print("Error: No structures found in the list file.")
return
# Load the average structure
cmd.load(average_file, 'average_structure')
# Prompt the user to define the residue range for alignment
residue_range = input("Enter the residue range for alignment (e.g., 6-77): ")
selection = f"resi {residue_range} and name CA+C+N+O"
# Align all structures to the average structure and save the ensemble
with open(output_ensemble, 'w') as output:
for i, structure_file in enumerate(structure_files):
structure_name = f'structure_{i+1}'
cmd.load(structure_file, structure_name)
# Align to the average structure
alignment_result = cmd.align(f'{structure_name} and ({selection})', f'average_structure and ({selection})')
print(f"Aligned {structure_file} to average structure with RMSD: {alignment_result[0]:.3f}")
# Save the aligned structure in MODEL/ENDMDL format
cmd.save(f'/tmp/aligned_{i+1}.pdb', structure_name)
with open(f'/tmp/aligned_{i+1}.pdb', 'r') as aligned_file:
output.write(f'MODEL {i+1}\n')
output.write(aligned_file.read())
output.write('ENDMDL\n')
print(f"Aligned ensemble saved as {output_ensemble}")
def compute_rmsd_to_average(list_file, average_file, rmsd_output_file):
"""
Compute RMSD of all structures to the average structure and save results to a text file.
Parameters:
list_file (str): Path to the list file containing structure file paths.
average_file (str): Path to the average structure PDB file.
rmsd_output_file (str): Path to save the RMSD results as a text file.
"""
# Initialize PyMOL without GUI
pymol.finish_launching(['pymol', '-qc'])
# Read the list of PDB files
with open(list_file, 'r') as f:
structure_files = [line.strip() for line in f if line.strip()]
if not structure_files:
print("Error: No structures found in the list file.")
return
# Load the average structure
cmd.load(average_file, 'average_structure')
# Prompt the user to define the residue range for RMSD calculation
residue_range = input("Enter the residue range for RMSD calculation (e.g., 6-77): ")
selection_backbone = f"resi {residue_range} and name CA+C+N+O"
# Compute RMSD for all structures
all_residues_rmsd = []
all_atoms_rmsd = []
backbone_rmsd = []
with open(rmsd_output_file, 'w') as rmsd_output:
rmsd_output.write("Structure\tAll_Residues_RMSD\tAll_Atoms_RMSD\tBackbone_RMSD\n")
for i, structure_file in enumerate(structure_files):
structure_name = f'structure_{i+1}'
cmd.load(structure_file, structure_name)
# Align and compute RMSD for all residues and atoms
rmsd_all_residues = cmd.align(f'{structure_name}', 'average_structure')[0]
rmsd_all_atoms = cmd.rms_cur(f'{structure_name}', 'average_structure')
rmsd_backbone = cmd.align(f'{structure_name} and ({selection_backbone})', f'average_structure and ({selection_backbone})')[0]
all_residues_rmsd.append(rmsd_all_residues)
all_atoms_rmsd.append(rmsd_all_atoms)
backbone_rmsd.append(rmsd_backbone)
rmsd_output.write(f"{structure_file}\t{rmsd_all_residues:.3f}\t{rmsd_all_atoms:.3f}\t{rmsd_backbone:.3f}\n")
print(f"Structure {structure_file} - All residues RMSD: {rmsd_all_residues:.3f}, All atoms RMSD: {rmsd_all_atoms:.3f}, Backbone RMSD: {rmsd_backbone:.3f}")
# Calculate overall RMSD values
avg_all_residues_rmsd = np.mean(all_residues_rmsd)
avg_all_atoms_rmsd = np.mean(all_atoms_rmsd)
avg_backbone_rmsd = np.mean(backbone_rmsd)
# Append overall RMSD values to the output file
with open(rmsd_output_file, 'a') as rmsd_output:
rmsd_output.write("\nOverall RMSD:\n")
rmsd_output.write(f"All residues: {avg_all_residues_rmsd:.3f}\n")
rmsd_output.write(f"All atoms: {avg_all_atoms_rmsd:.3f}\n")
rmsd_output.write(f"Backbone: {avg_backbone_rmsd:.3f}\n")
print(f"Overall RMSD (backbone for all residues): {avg_all_residues_rmsd:.3f}")
print(f"Overall RMSD (all atoms): {avg_all_atoms_rmsd:.3f}")
print(f"Overall RMSD (backbone of user defined range): {avg_backbone_rmsd:.3f}")
# Cleanup PyMOL session
cmd.quit()
if __name__ == "__main__":
# Input and output file paths
list_file = "List_best20" # Replace with your list file
average_file = "average.pdb" # Replace with your average file
output_ensemble = "aligned_ensemble.pdb" # Replace with your desired output ensemble file
rmsd_output_file = "rmsd_results.txt" # Replace with your desired RMSD output file
# Generate the average structure
generate_average_structure(list_file, average_file)
# Align structures to the average structure and save ensemble
align_to_average(list_file, average_file, output_ensemble)
print(f"Aligned ensemble generated and saved to {output_ensemble}.")
# Compute RMSD to average structure and save results
compute_rmsd_to_average(list_file, average_file, rmsd_output_file)
print(f"RMSD results saved to {rmsd_output_file}.")
The output will be similar with the example of our in-house ubiquitin NMR data here:
shell> python align_and_ensemble.py
Average structure saved as average.pdb
Enter the residue range for alignment (e.g., 6-77): 6-77
Aligned UbRefine_89.pdb to average structure with RMSD: 0.469
Aligned UbRefine_17.pdb to average structure with RMSD: 0.340
Aligned UbRefine_27.pdb to average structure with RMSD: 0.297
Aligned UbRefine_65.pdb to average structure with RMSD: 0.425
Aligned UbRefine_98.pdb to average structure with RMSD: 0.428
Aligned UbRefine_74.pdb to average structure with RMSD: 0.436
Aligned UbRefine_68.pdb to average structure with RMSD: 0.372
Aligned UbRefine_83.pdb to average structure with RMSD: 0.327
Aligned UbRefine_19.pdb to average structure with RMSD: 0.415
Aligned UbRefine_45.pdb to average structure with RMSD: 0.378
Aligned UbRefine_78.pdb to average structure with RMSD: 0.371
Aligned UbRefine_67.pdb to average structure with RMSD: 0.375
Aligned UbRefine_1.pdb to average structure with RMSD: 0.275
Aligned UbRefine_24.pdb to average structure with RMSD: 0.406
Aligned UbRefine_92.pdb to average structure with RMSD: 0.249
Aligned UbRefine_18.pdb to average structure with RMSD: 0.300
Aligned UbRefine_63.pdb to average structure with RMSD: 0.498
Aligned UbRefine_82.pdb to average structure with RMSD: 0.398
Aligned UbRefine_28.pdb to average structure with RMSD: 0.413
Aligned UbRefine_99.pdb to average structure with RMSD: 0.557
Aligned ensemble saved as aligned_ensemble.pdb
Aligned ensemble generated and saved to aligned_ensemble.pdb.
Enter the residue range for RMSD calculation (e.g., 6-77): 6-77
Structure UbRefine_89.pdb - All residues RMSD: 0.839, All atoms RMSD: 1.976, Backbone RMSD: 0.469
Structure UbRefine_17.pdb - All residues RMSD: 0.714, All atoms RMSD: 2.008, Backbone RMSD: 0.339
Structure UbRefine_27.pdb - All residues RMSD: 0.708, All atoms RMSD: 1.751, Backbone RMSD: 0.295
Structure UbRefine_65.pdb - All residues RMSD: 0.817, All atoms RMSD: 2.405, Backbone RMSD: 0.425
Structure UbRefine_98.pdb - All residues RMSD: 0.771, All atoms RMSD: 1.758, Backbone RMSD: 0.428
Structure UbRefine_74.pdb - All residues RMSD: 0.878, All atoms RMSD: 1.981, Backbone RMSD: 0.436
Structure UbRefine_68.pdb - All residues RMSD: 0.723, All atoms RMSD: 2.511, Backbone RMSD: 0.374
Structure UbRefine_83.pdb - All residues RMSD: 0.696, All atoms RMSD: 1.820, Backbone RMSD: 0.327
Structure UbRefine_19.pdb - All residues RMSD: 0.883, All atoms RMSD: 2.442, Backbone RMSD: 0.416
Structure UbRefine_45.pdb - All residues RMSD: 0.755, All atoms RMSD: 1.892, Backbone RMSD: 0.377
Structure UbRefine_78.pdb - All residues RMSD: 0.801, All atoms RMSD: 1.832, Backbone RMSD: 0.372
Structure UbRefine_67.pdb - All residues RMSD: 0.771, All atoms RMSD: 1.817, Backbone RMSD: 0.375
Structure UbRefine_1.pdb - All residues RMSD: 0.752, All atoms RMSD: 2.114, Backbone RMSD: 0.273
Structure UbRefine_24.pdb - All residues RMSD: 0.829, All atoms RMSD: 2.094, Backbone RMSD: 0.405
Structure UbRefine_92.pdb - All residues RMSD: 0.728, All atoms RMSD: 1.668, Backbone RMSD: 0.250
Structure UbRefine_18.pdb - All residues RMSD: 0.726, All atoms RMSD: 1.813, Backbone RMSD: 0.300
Structure UbRefine_63.pdb - All residues RMSD: 0.852, All atoms RMSD: 2.038, Backbone RMSD: 0.495
Structure UbRefine_82.pdb - All residues RMSD: 0.816, All atoms RMSD: 1.630, Backbone RMSD: 0.397
Structure UbRefine_28.pdb - All residues RMSD: 0.692, All atoms RMSD: 1.999, Backbone RMSD: 0.412
Structure UbRefine_99.pdb - All residues RMSD: 1.012, All atoms RMSD: 2.030, Backbone RMSD: 0.557
Overall RMSD (backbone for all residues): 0.788
Overall RMSD (all atoms): 1.979
Overall RMSD (backbone of user defined range): 0.386
RMSD results saved to rmsd_results.txt.The average structure of ubiquitin from 20 refined lowest-energy structures
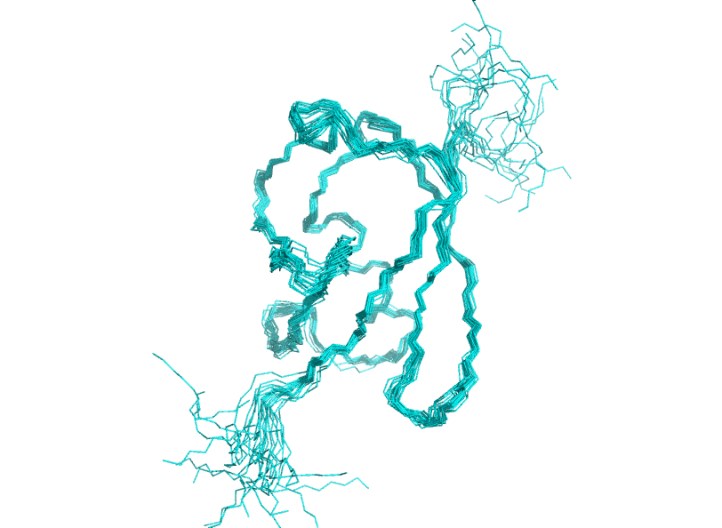
Directly superimpose 20 conformers which aligned by Xplor-NIH previously. As this is a full-length based alignment, the disordered tails drive the superposition quality obviously.
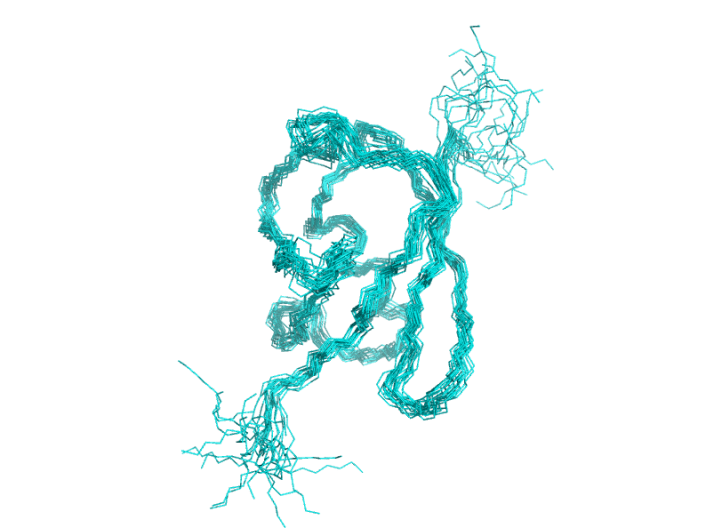
I aligned 6-77 (residues 1-5 are extra N-terminal residues GSGGS) using this python script, it shows a much better alignment. These 20 conformers are no showing a great agreement in the secondary structural regions, and very dynamic in the N- (top) and C-(bottom) termini.